
GOLD Report: Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease
Publication date: 15

Chapter 1: Definition and Overview
Chronic Obstructive Pulmonary Disease (COPD) is a common, preventable and treatable disease that is characterized by persistent respiratory symptoms and airflow limitation that is due to airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases and influenced by host factors including abnormal lung development. Significant comorbidities may have an impact on morbidity and mortality.
There may be significant lung pathology (e.g., emphysema) in the absence of airflow limitation that needs further evaluation (Figure 1.1). The chronic airflow limitation that is characteristic of COPD is caused by a mixture of small airways disease and parenchymal destruction (emphysema), the relative contributions of which vary from person to person. These changes do not always occur together, but evolve at different rates over time. Chronic inflammation causes structural changes, narrowing of the small airways and destruction of the lung parenchyma that leads to the loss of alveolar attachments to the small airways and decreases lung elastic recoil. In turn, these changes diminish the ability of the airways to remain open during expiration. A loss of small airways may also contribute to airflow limitation and mucociliary dysfunction is a characteristic feature of the disease. Airflow limitation is usually measured by spirometry as this is the most widely available and reproducible test of lung function. Many previous definitions of COPD have emphasized the terms “emphysema” and “chronic bronchitis”, which are not included in the definition used in this or earlier GOLD reports. Emphysema, or destruction of the gas-exchanging surfaces of the lung (alveoli), is a pathological term that is often (but incorrectly) used clinically and describes only one of several structural abnormalities present in patients with COPD. Chronic bronchitis, or the presence of cough and sputum production for at least 3 months in each of two consecutive years, remains a clinically and epidemiologically useful term, but is present in only a minority of subjects when this definition is used. However, when alternative definitions are used to define chronic bronchitis, or older populations with greater levels of smoke or occupational inhalant exposure are queried, the prevalence of chronic bronchitis is greater.1,2 It is important to recognize that chronic respiratory symptoms may precede the development of airflow limitation and may be associated with the development of acute respiratory events.3 Chronic respiratory symptoms also exist in individuals with normal spirometry3,4 and a significant number of smokers without airflow limitation have structural evidence of lung disease manifested by the varying presence of emphysema, airway wall thickening and gas trapping.3,4
COPD is a leading cause of morbidity and mortality worldwide that induces an economic and social burden that is both substantial and increasing.5,6 COPD prevalence, morbidity and mortality vary across countries and across different groups within countries. COPD is the result of a complex interplay of long-term cumulative exposure to noxious gases and particles, combined with a variety of host factors including genetics, airway hyper-responsiveness and poor lung growth during childhood.7-9 Often, the prevalence of COPD is directly related to the prevalence of tobacco smoking, although in many countries outdoor, occupational and indoor air pollution (resulting from the burning of wood and other biomass fuels) are major COPD risk factors.10,11 The prevalence and burden of COPD are projected to increase over the coming decades due to continued exposure to COPD risk factors and aging of the world’s population; as longevity increases more people will express the long-term effects of exposure to COPD risk factors.12 Information on the burden of COPD can be found on international websites, for example the:
Existing COPD prevalence data vary widely due to differences in survey methods, diagnostic criteria, and analytical approaches.12 Importantly, all of the studies defined COPD by spirometry alone and not by the combination of symptoms and spirometry. The lowest estimates of prevalence are those based on self-reporting of a doctor’s diagnosis of COPD, or equivalent condition. For example, most national data show that < 6% of the adult population have been told that they have COPD.15 This is likely to be a reflection of the widespread under-recognition and under-diagnosis of COPD.16
Despite the complexities, data are emerging that enable more accurate estimates of COPD prevalence. A systematic review and meta-analysis, including studies carried out in 28 countries between 1990 and 2004,15 provided evidence that the prevalence of COPD is appreciably higher in smokers and ex-smokers compared to non-smokers, in those ≥ 40 years of age compared to those < 40, and in men compared to women. The Latin American Project for the Investigation of Obstructive Lung Disease (PLATINO)17 examined the prevalence of post-bronchodilator airflow limitation among persons > 40 years in one major city from each of five Latin American countries – Brazil, Chile, Mexico, Uruguay, and Venezuela. In each country, the prevalence of COPD increased steeply with age, with the highest prevalence among those > 60 years. Prevalence in the total population ranged from a low of 7.8% in Mexico City, Mexico, to a high of 19.7% in Montevideo, Uruguay. In all five cities, the prevalence was appreciably higher in men than in women,17 which contrasts with findings from European cities such as Salzburg, Austria.18
The Burden of Obstructive Lung Diseases (BOLD) program has also used a standardized methodology comprising questionnaires and pre- and post-bronchodilator spirometry to assess the prevalence and risk factors for COPD in people aged 40 and over around the world. Surveys have been completed in 29 countries and studies are on-going in a further nine.19 BOLD reported worse lung function than earlier studies, with a prevalence of COPD grade 2 or higher of 10.1% (SE 4.8) overall, 11.8% (SE 7.9) for men, and 8.5% (SE 5.8) for women20 and a substantial prevalence of COPD of 3-11% among never-smokers.20 BOLD also examined the prevalence of COPD in north and sub-Saharan Africa and Saudi Arabia and found similar results.21-24
Based on BOLD and other large scale epidemiological studies, it is estimated that the number of COPD cases was 384 million in 2010, with a global prevalence of 11.7% (95% confidence interval (CI) 8.4%–15.0%).25 Globally, there are around three million deaths annually.26 With the increasing prevalence of smoking in developing countries, and aging populations in high-income countries, the prevalence of COPD is expected to rise over the next 40 years and by 2060 there may be over 5.4 million deaths annually from COPD and related conditions.27,28 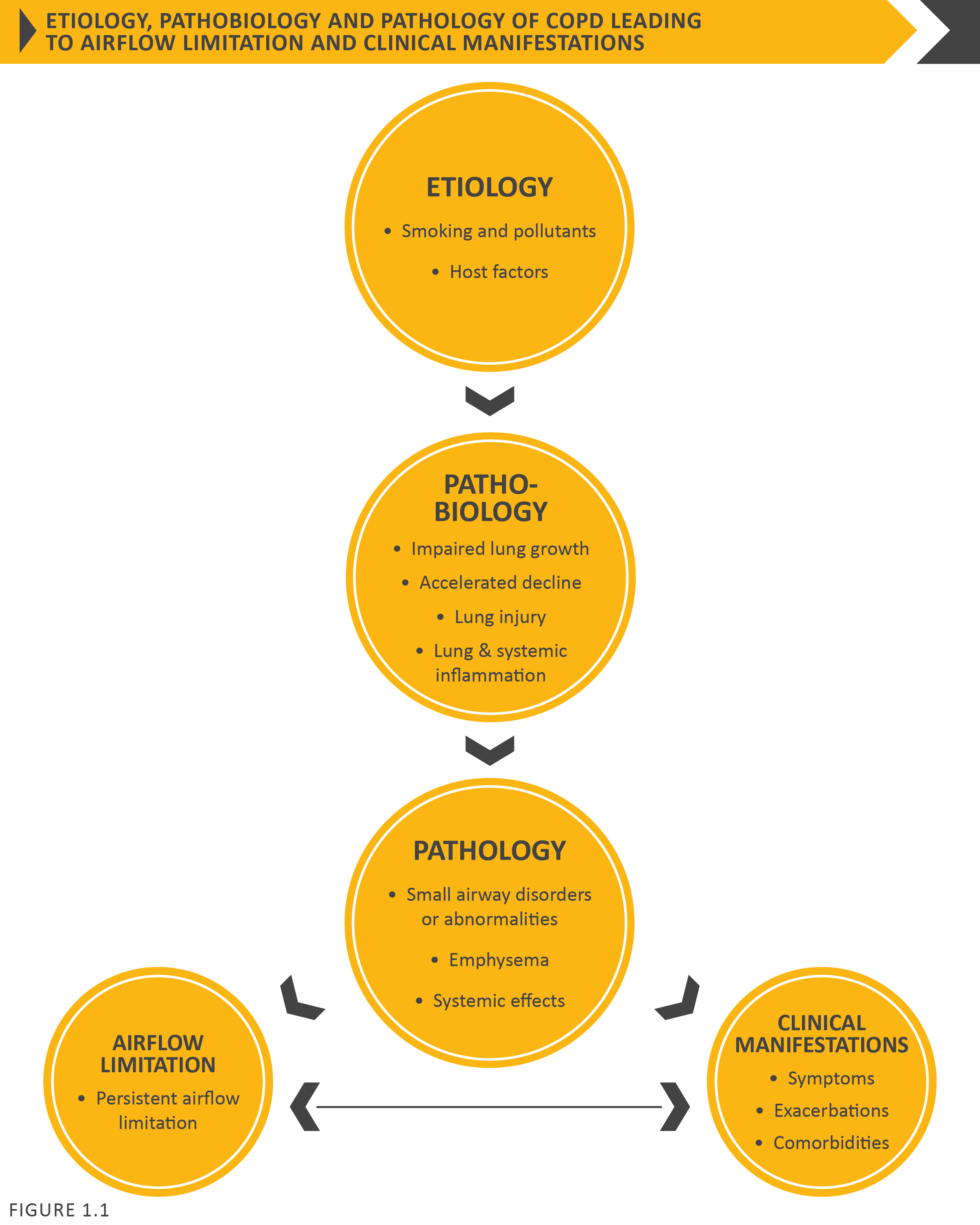
Morbidity measures traditionally include physician visits, emergency department visits, and hospitalizations. Although COPD databases for these outcome parameters are less readily available and usually less reliable than mortality databases, to date studies on the available data indicate that morbidity due to COPD increases with age,15-17 and in patients with COPD the development of comorbidities may be seen at an earlier age.29 Morbidity from COPD may be affected by other concomitant chronic conditions (e.g., cardiovascular disease,30 musculoskeletal impairment, diabetes mellitus) that are related to smoking, aging and COPD. These chronic conditions may significantly impair patient’s health status, in addition to interfering with COPD management and are major drivers of hospitalizations and costs for patients with COPD.31
The World Health Organization (WHO) publishes mortality statistics for selected causes of death annually for all WHO regions; additional information is available from the WHO Evidence for Health Policy Department.32 However, data must be interpreted with caution because of the inconsistent use of COPD terminology. In the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD-10), deaths from COPD or chronic airways obstruction are included in the broad category of “COPD and allied conditions” (ICD-10 codes J42-46).
Under-recognition and under-diagnosis of COPD reduces the accuracy of mortality data.33,34 Furthermore, the accuracy of COPD diagnosis codes recorded in administrative health databases is also uncertain.35,36 In some jurisdictions, reliance on administrative health data, particularly those that only record hospitalizations, may underestimate the burden of COPD.37 The reliability of recording of COPD-related deaths in mortality data is also problematic. Although COPD is often a primary cause of death, it is more likely to be listed as a contributory cause of death or omitted from the death certificate entirely.38 However, it is clear that COPD is one of the most important causes of death in most countries. For instance, in 2011, COPD was the third leading cause of death in the United States.39 This increase in COPD-related mortality has mainly been driven by the expanding epidemic of smoking; reduced mortality from other common causes of death (e.g., ischemic heart disease, infectious diseases); the aging of the world’s population, particularly in high-income countries; and scarcity of effective disease modifying therapies.
COPD is associated with significant economic burden. In the European Union, the total direct costs of respiratory disease are estimated to be about 6% of the total annual healthcare budget, with COPD accounting for 56% (38.6 billion Euros) of the cost of respiratory disease.40 In the United States the estimated direct costs of COPD are $32 billion and the indirect costs $20.4 billion.41 COPD exacerbations account for the greatest proportion of the total COPD burden on the healthcare system. Not surprisingly, there is a striking direct relationship between the severity of COPD and the cost of care, and the cost distribution changes as the disease progresses. For example, hospitalization and ambulatory oxygen costs soar as COPD severity increases. Any estimate of direct medical expenditure for home-based care under-represents the true cost of home-based care to society, because it ignores the economic value of the care provided by family members to people with COPD.
In developing countries, direct medical costs may be less important than the impact of COPD on workplace and home productivity. Because the healthcare sector might not provide long-term supportive care services for severely disabled individuals, COPD may force at least two individuals to leave the workplace – the affected individual and a family member who must now stay home to care for their disabled relative.42 Since human capital is often the most important national asset for developing countries, the indirect costs of COPD may represent a serious threat to the economy.
Since mortality offers only a limited perspective on the human burden of a disease, it is desirable to find other measures of disease burden that are consistent and measurable within and between nations. The authors of the Global Burden of Disease (GBD) Study designed a method to estimate the fraction of mortality and disability attributable to major diseases and injuries using a composite measure of the burden of each health problem: the Disability-Adjusted Life Year (DALY).43 The DALYs for a specific condition are the sum of years lost because of premature mortality and years of life lived with disability, adjusted for the severity of disability. The GBD Study found that COPD is an increasing contributor to disability and mortality around the world. In 2005 COPD was the eighth leading cause of DALYs lost across the world but by 2013 COPD was ranked as the fifth leading cause of DALYs lost.44 In the United States, COPD is the second leading cause of reduced DALYs, trailing only ischemic heart disease.45
Although cigarette smoking is the most well studied COPD risk factor, it is not the only risk factor and there is consistent evidence from epidemiologic studies that non-smokers may also develop chronic airflow limitation.20 Much of the evidence concerning risk factors for COPD comes from cross-sectional epidemiological studies that identify associations rather than causal relationships. Nevertheless, compared to smokers with COPD, never smokers with chronic airflow limitation have fewer symptoms, milder disease and lower burden of systemic inflammation.46Interestingly, never smokers with chronic airflow limitation do not appear to have an increased risk of lung cancer, or cardiovascular comorbidities, compared to those without chronic airflow limitation. However, there is evidence that they have an increased risk of pneumonia and mortality from respiratory failure.46
Although several longitudinal studies of COPD have followed groups and populations for up to 20 years,7 to date no studies have monitored the progression of the disease through its entire course, or included the pre and perinatal periods that may be important in shaping an individual’s future COPD risk. Thus, the current understanding of risk factors for COPD is in many respects still incomplete.
COPD results from a complex interaction between genes and the environment. Cigarette smoking is the leading environmental risk factor for COPD, yet even for heavy smokers, fewer than 50% develop COPD during their lifetime.47 Although genetics may play a role in modifying the risk of COPD in smokers, there may also be other risk factors involved. For example, sex may influence whether a person takes up smoking or experiences certain occupational or environmental exposures; socioeconomic status may be linked to a child’s birth weight (as it impacts on lung growth and development, and in turn on susceptibility to developing the disease); and longer life expectancy will allow greater lifetime exposure to risk factors. Understanding the relationships and interactions between risk factors requires further investigation.
The genetic risk factor that is best documented is a severe hereditary deficiency of alpha-1 antitrypsin (AATD),48 a major circulating inhibitor of serine proteases. Although AATD deficiency is relevant to only a small part of the world’s population, it illustrates the interaction between genes and environmental exposures that predispose an individual to COPD. A systematic review of 20 studies in European populations found AATD PiZZ genotypes in 0.12% of COPD patients (range 0.08-0.24%), and a prevalence ranging from 1 in 408 in Northern Europe to 1 in 1,274 in Eastern Europe.49
A significant familial risk of airflow limitation has been observed in people who smoke and are siblings of patients with severe COPD,50 suggesting that genetics together with environmental factors could influence this susceptibility. Single genes, such as the gene encoding matrix metalloproteinase 12 (MMP-12) and glutathione S-transferase have been related to a decline in lung function51 or risk of COPD.52 Several genome-wide association studies have linked genetic loci with COPD (or FEV1 or FEV1/FVC as the phenotype) including markers near the alpha-nicotinic acetylcholine receptor, hedgehog interacting protein (HHIP), and several others. Nevertheless, it remains uncertain whether these genes are directly responsible for COPD or are merely markers of causal genes.53-57
Age is often listed as a risk factor for COPD. It is unclear if healthy aging as such leads to COPD or if age reflects the sum of cumulative exposures throughout life.58 Aging of the airways and parenchyma mimic some of the structural changes associated with COPD.58 Gender related differences in immune pathways and pattern of airway damage may be seen and might be clinically important. More work in this area is needed. In the past, most studies have reported that COPD prevalence and mortality are greater among men than women, but later data from developed countries has reported that the prevalence of COPD is now almost equal in men and women, probably reflecting the changing patterns of tobacco smoking.59 Although controversial, some studies have even suggested that women are more susceptible to the effects of tobacco smoke than men,60-62 leading to more severe disease for the equivalent quantity of cigarettes consumed. This notion has been validated in animal studies and human pathology specimens, which have demonstrated a greater burden of small airway disease in females compared with males with COPD despite a similar history of tobacco smoke exposure.63,64
Processes occurring during gestation, birth, and exposures during childhood and adolescence affect lung growth.65,66Reduced maximal attained lung function (as measured by spirometry) may identify individuals who are at increased risk for the development of COPD.4,8 Any factor that affects lung growth during gestation and childhood has the potential for increasing an individual’s risk of developing COPD. For example, a large study and meta-analysis confirmed a positive association between birthweight and FEV1 in adulthood,67 and several studies have found an effect of early childhood lung infections. Factors in early life termed “childhood disadvantage factors” seem to be as important as heavy smoking in predicting lung function in adult life.67 One study evaluated three different longitudinal cohorts and found that approximately 50% of patients developed COPD due to accelerated decline in FEV1 over time, while the other 50% developed COPD due to abnormal lung growth and development (with normal decline in lung function over time; Figure 1.2).7 The Medical Research Council National Survey of Health and Development documented a synergistic interaction between smoking and infant respiratory infection as well as early life home overcrowding with lung function at age 43.68
Across the world, cigarette smoking is the most commonly encountered risk factor for COPD. Cigarette smokers have a higher prevalence of respiratory symptoms and lung function abnormalities, a greater annual rate of decline in FEV1, and a greater COPD mortality rate than non-smokers.69 Other types of tobacco (e.g., pipe, cigar, water pipe)70-72 and marijuana73 are also risk factors for COPD. Passive exposure to cigarette smoke, also known as environmental tobacco smoke (ETS), may also contribute to respiratory symptoms and COPD74 by increasing the lung’s total burden of inhaled particles and gases. Smoking during pregnancy may pose a risk for the fetus, by affecting lung growth and development in utero, and possibly the priming of the immune system.75
Occupational exposures, including organic and inorganic dusts, chemical agents and fumes, are an under-appreciated risk factor for COPD.10,76 A study of the population-based UK biobank cohort identified occupations including sculptors, gardeners and warehouse workers that were associated with an increased COPD risk among never-smokers and never-asthmatics.77 A cross-sectional observational study demonstrated that self-reported exposure to workplace dust and fumes is not only associated with increased airflow limitation and respiratory symptoms, but also with more emphysema and gas trapping, assessed by computed tomography scan, in both men and women.78 An analysis of the large U.S. population-based National Health and Nutrition Examination Survey III survey of almost 10,000 adults aged 30-75 years estimated the fraction of COPD attributable to workplace exposures was 19.2% overall, and 31.1% among never-smokers.79 These estimates are consistent with a statement published by the American Thoracic Society that concluded that occupational exposures account for 10-20% of either symptoms or functional impairment consistent with COPD.80 The risk from occupational exposures in less regulated areas of the world is likely to be much higher than reported in studies from Europe and North America.
Wood, animal dung, crop residues, and coal, typically burned in open fires or poorly functioning stoves, may lead to very high levels of indoor air pollution.81 There is growing evidence that indoor biomass exposure to modern and traditional fuels used during cooking may predispose women to develop COPD in many developing countries.82-85Almost three billion people worldwide use biomass and coal as their main source of energy for cooking, heating, and other household needs, so the population at risk worldwide is very large.86,87 There is a lack of research about biomass related COPD,88 although there is limited evidence from an observational study that switching to cleaner cooking fuels or reducing exposure may reduce COPD risk in non-smokers.89
High levels of urban air pollution are harmful to individuals with existing heart or lung disease. The role of outdoor air pollution as a risk factor for COPD is unclear, but its role appears to be relatively small in adults compared to the role of cigarette smoking.10 Cross-sectional analyses have shown an association between ambient levels of particulate matter (PM2.5/10) and COPD prevalence.90,91 However, there is evidence that air pollution has a significant impact on lung maturation and development. For instance, the Children’s Health Study found that children from communities with the highest levels of outdoor nitrogen dioxide (NO2) and particulate matter < 2.5 μm in aerodynamic diameter (PM2.5) were nearly 5 times more likely to have reduced lung function (defined as FEV1 < 80% of predicted) compared to children from communities with the lowest levels of NO2 and PM2.5.92 Importantly, reduction in ambient NO2 and PM2.5 levels significantly mitigated the risk of experiencing impaired lung growth.93 However, the relative effects of short-term, high-peak exposures and long-term, low-level exposures are yet to be resolved.
Poverty is consistently associated with airflow obstruction94 and lower socioeconomic status is associated with an increased risk of developing COPD.95,96 It is not clear, however, whether this pattern reflects exposures to indoor and outdoor air pollutants, crowding, poor nutrition, infections, or other factors related to low socioeconomic status.
Asthma may be a risk factor for the development of chronic airflow limitation and COPD. In a report from a longitudinal cohort of the Tucson Epidemiological Study of Airway Obstructive Disease, adults with asthma were found to have a 12-fold higher risk of acquiring COPD over time compared to those without asthma, after adjusting for smoking.97Another longitudinal study of people with asthma found that around 20% of subjects developed irreversible airflow limitation and reduced transfer coefficient.98 A third longitudinal study observed that self-reported asthma was associated with excess loss of FEV1 in the general population.99 A study examining the pattern of lung-growth decline in children with asthma found that 11% met lung function impairment consistent with the spirometric classification of COPD in early adulthood.100 In the European Community Respiratory Health Survey, airway hyper-responsiveness was second only to cigarette smoking as the leading risk factor for COPD, responsible for 15% of the population attributable risk (smoking had a population attributable risk of 39%).101 The pathology of chronic airflow limitation in asthmatic non-smokers and non-asthmatic smokers is markedly different, suggesting that the two disease entities may remain different even when presenting with similarly reduced lung function.97,103 However, separating asthma from COPD in adults may be clinically difficult at times.
Airway hyper-responsiveness can exist without a clinical diagnosis of asthma and has been shown to be an independent predictor of COPD and respiratory mortality in population studies104,105 as well as an indicator of risk of excess decline in lung function in patients with mild COPD.106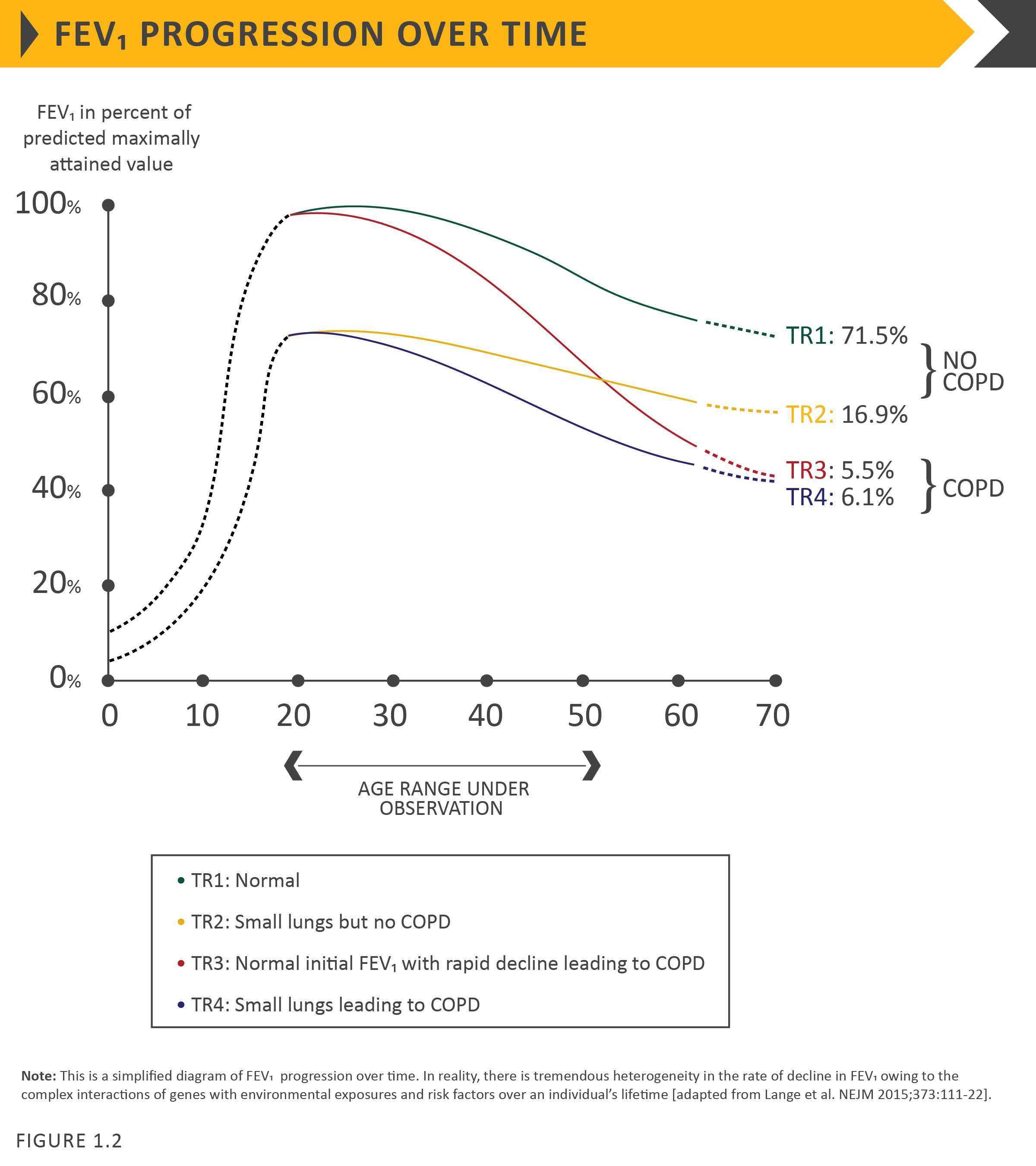
In the seminal study by Fletcher and colleagues, chronic bronchitis was not associated with an accelerated decline in lung function.96,107 However, subsequent studies have observed an association between mucus hypersecretion and increased FEV1 decline,108 and in younger adults who smoke, the presence of chronic bronchitis has been associated with an increased likelihood of developing COPD.109 Chronic bronchitis has also been associated with an increased risk in the total number as well as severity of exacerbations.110
A history of severe childhood respiratory infection has been associated with reduced lung function and increased respiratory symptoms in adulthood.101 Susceptibility to infections plays a role in exacerbations of COPD but the effect on disease development is less clear. In a large observational study Pseudomonas aeruginosa colonization independently predicted an increased risk of hospitalization for exacerbation and all-cause mortality.111 There is evidence that HIV patients are at increased risk of COPD compared to HIV negative controls (11 studies; pooled odds ratio for 1.14 (95% CI 1.05,1.25)112; tuberculosis has also been identified as a risk factor for COPD.113 In addition, tuberculosis is both a differential diagnosis for COPD and a potential comorbidity.114,115
Inhalation of cigarette smoke or other noxious particles, such as smoke from biomass fuels, causes lung inflammation. Lung inflammation is a normal response that appears to be modified in patients who develop COPD. This chronic inflammatory response may induce parenchymal tissue destruction (resulting in emphysema), and disruption of normal repair and defense mechanisms (resulting in small airway fibrosis). These pathological changes lead to gas trapping and progressive airflow limitation. A brief overview follows that describes and summarizes the pathologic changes in COPD, their cellular and molecular mechanisms, and how these underlie the physiological abnormalities and symptoms characteristic of this disease.
Pathological changes characteristic of COPD are found in the airways, lung parenchyma, and pulmonary vasculature.116 The pathological changes observed in COPD include chronic inflammation, with increased numbers of specific inflammatory cell types in different parts of the lung, and structural changes resulting from repeated injury and repair. In general, the inflammatory and structural changes in the airways increase with disease severity and persist on smoking cessation. Most pathology data come from studies in smokers and the same balance of airway and parenchymal disease cannot necessarily be assumed when other factors are operative. Systemic inflammation may be present and could play a role in the multiple comorbid conditions found in patients with COPD.117
The inflammation observed in the respiratory tract of COPD patients appears to be a modification of the normal inflammatory response of the respiratory tract to chronic irritants such as cigarette smoke. The mechanisms for this amplified inflammation are not yet understood but may, at least in part, be genetically determined. Although some patients develop COPD without smoking, the nature of the inflammatory response in these patients is as yet unknown. Oxidative stress and an excess of proteinases in the lung are likely to further modify lung inflammation. Together, these mechanisms may lead to the characteristic pathological changes in COPD. Lung inflammation persists after smoking cessation through unknown mechanisms, although autoantigens and perturbations in the lung microbiome may play a role.118,119 Similar mechanisms may occur for concomitant chronic diseases.
Oxidative stress. Oxidative stress may be an important amplifying mechanism in COPD.117,120 Biomarkers of oxidative stress (e.g., hydrogen peroxide, 8-isoprostane) are increased in the exhaled breath condensate, sputum, and systemic circulation of COPD patients. Oxidative stress is further increased during exacerbations. Oxidants are both generated by cigarette smoke and other inhaled particulates, and released from activated inflammatory cells such as macrophages and neutrophils. There may also be a reduction in endogenous antioxidants in COPD patients as a result of reduction in levels of the transcription factor Nrf2 that regulates many antioxidant genes.114,121
Protease-antiprotease imbalance. There is compelling evidence for an imbalance in the lungs of COPD patients between proteases that break down connective tissue components and antiproteases that counterbalance this action.122 Increased levels of several proteases, derived from inflammatory cells and epithelial cells, have been observed in COPD patients. There is increasing evidence that these proteases may interact with each other. Protease-mediated destruction of elastin, a major connective tissue component in lung parenchyma, is believed to be an important feature of emphysema but may be more difficult to establish in airway changes.123
Inflammatory cells. COPD is characterized by increased numbers of macrophages in peripheral airways, lung parenchyma and pulmonary vessels, together with increased activated neutrophils and increased lymphocytes that include Tc1, Th1, Th17 and ILC3 cells. In some patients, there may also be increases in eosinophils, Th2 or ILC2 cells. All of these inflammatory cells, together with epithelial cells and other structural cells release multiple inflammatory mediators.117 A study suggests that local IgA deficiency is associated with bacterial translocation, small airway inflammation and airway remodeling.124
Inflammatory mediators. The wide variety of inflammatory mediators that have been shown to be increased in COPD patients attract inflammatory cells from the circulation (chemotactic factors), amplify the inflammatory process (proinflammatory cytokines), and induce structural changes (growth factors).125
Peribronchiolar and interstitial fibrosis. Peribronchiolar fibrosis and interstitial opacities have been reported in patients with COPD or those who are asymptomatic smokers.118,126-128 An excessive production of growth factors may be found in smokers or those with preceding airway inflammation who have COPD.129 Inflammation may precede the development of fibrosis or repeated injury of the airway wall itself may lead to excessive production of muscle and fibrous tissue.130 This may be a contributing factor to the development of small airways limitation and eventually the obliteration that may precede the development of emphysema.131
Differences in inflammation between COPD and asthma. Although both COPD and asthma are associated with chronic inflammation of the respiratory tract, there are differences in the inflammatory cells and mediators involved in the two diseases.132 Some patients with COPD have an inflammatory pattern with increased eosinophils.133
There is now a good understanding of how the underlying disease process in COPD leads to the characteristic physiological abnormalities and symptoms. For example, inflammation and narrowing of peripheral airways leads to decreased FEV1.134 Parenchymal destruction due to emphysema also contributes to airflow limitation and leads to decreased gas transfer. There is also emerging evidence to suggest that in addition to airway narrowing, there is a loss of small airways, which may contribute to airflow limitation.135
Airflow limitation and gas trapping. The extent of inflammation, fibrosis, and luminal exudates in the small airways correlates with the reduction in the FEV1 and FEV1/FVC ratio, and probably with the accelerated decline in FEV1 that is characteristic of COPD.134 This peripheral airway limitation progressively traps gas during expiration, resulting in hyperinflation. Static hyperinflation reduces inspiratory capacity and is commonly associated with dynamic hyperinflation during exercise leading to increased dyspnea and limitation of exercise capacity. These factors contribute to impairment of the intrinsic contractile properties of respiratory muscles. It is thought that hyperinflation develops early in the disease and is the main mechanism for exertional dyspnea.136,137 Bronchodilators acting on peripheral airways reduce gas trapping, thereby reducing lung volumes and improving symptoms and exercise capacity.138
Gas exchange abnormalities. Gas exchange abnormalities result in hypoxemia and hypercapnia, and have several mechanisms in COPD. In general, gas transfer for oxygen and carbon dioxide worsens as the disease progresses. Reduced ventilation may also be due to reduced ventilatory drive or increased dead space ventilation.137 This may lead to carbon dioxide retention when it is combined with reduced ventilation, due to increased effort to breathe because of severe limitation and hyperinflation coupled with ventilatory muscle impairment. The abnormalities in alveolar ventilation and a reduced pulmonary vascular bed further worsen the VA/Q (ventilation perfusion ratio) abnormalities.139
Mucus hypersecretion. Mucus hypersecretion, resulting in a chronic productive cough, is a feature of chronic bronchitis and is not necessarily associated with airflow limitation. Conversely, not all patients with COPD have symptomatic mucus hypersecretion. When present, mucus hypersecretion is due to an increased number of goblet cells and enlarged submucosal glands, both because of chronic airway irritation by cigarette smoke and other noxious agents. Several mediators and proteases stimulate mucus hypersecretion and many of them exert their effects through the activation of epidermal growth factor receptor (EGFR).140
Pulmonary hypertension. Pulmonary hypertension may develop late in the course of COPD and is due mainly to hypoxic vasoconstriction of the small pulmonary arteries, eventually resulting in structural changes that include intimal hyperplasia and later smooth muscle hypertrophy/hyperplasia.141 Even in mild COPD, or in smokers susceptible to emphysema,142,143 there are significant abnormalities in pulmonary microvascular blood flow, that worsen with disease progression.144
An inflammatory response in vessels, similar to that seen in the airways, is also observed in COPD, along with evidence of endothelial cell dysfunction. The loss of the pulmonary capillary bed in emphysema may further contribute to increased pressure in the pulmonary circulation. Progressive pulmonary hypertension may lead to right ventricular hypertrophy and eventually to right-side cardiac failure. Interestingly, the diameter of pulmonary artery as measured on computed tomography (CT) scans has been shown to relate to the risk of exacerbation, independent of previous history of exacerbations.145 This suggests that perturbations in pulmonary vasculature are major, but under-recognized, drivers of symptoms and exacerbations in COPD.
Exacerbations. Exacerbations of respiratory symptoms triggered by respiratory infections with bacteria or viruses (which may coexist), environmental pollutants, or unknown factors often occur in patients with COPD; a characteristic response with increased inflammation occurs during episodes of bacterial or viral infection. During exacerbations there is increased hyperinflation and gas trapping, with reduced expiratory flow, thus accounting for increased dyspnea.146 There is also worsening of VA/Q abnormalities that can result in hypoxemia.147 During exacerbations there is evidence of increased airway inflammation. Other conditions (pneumonia, thromboembolism, and acute cardiac failure) may mimic or aggravate an exacerbation of COPD.
Systemic features. Most patients with COPD have concomitant chronic diseases linked to the same risk factors i.e., smoking, aging, and inactivity, which may have a major impact on health status and survival.148 Airflow limitation and particularly hyperinflation affect cardiac function and gas exchange.146 Inflammatory mediators in the circulation may contribute to skeletal muscle wasting and cachexia, and may initiate or worsen comorbidities such as ischemic heart disease, heart failure, osteoporosis, normocytic anemia, diabetes, and metabolic syndrome.